REPFIND is a program to find clustered, exact repeats in nucleotide sequences. For each repeat cluster that it finds, it calculates a P-value, which indicates the probability of finding such a concentration of that particular repeat just by chance. Of the many possible clusters for each repeated word, REPFIND selects the one with the most significant P-value.
We are using REPFIND to study localization of messenger RNAs. These molecules sometimes contain signals in their 3' untranslated regions that specify how they are transported in the cell, prior to being translated. This process is essential for establishing body axes in embryogenesis, among other things. The localization signals often appear to consist of repeat clusters that can be detected by REPFIND.
Publication: JN Betley, MC Frith, JH Graber, S Choo, JO Deshler (2002). A ubiquitous and conserved signal for RNA localization in chordates. Current Biology, 12, 1756-61.
Download REPFIND on to your computer. This link leads to a program in the PERL language implementing the REPFIND algorithm. You will need to have PERL installed on your computer, and for full functionality you will also need the program DUST which detects low complexity sequences. After downloading the program you need to ensure that it has execute permission (by entering the command 'chmod +x repfind' on Unix systems), and then you can obtain a help message with 'repfind -h'.
Return to Zlab Gene Regulation Hub
3-9-2004: Fixed bug in downloadable REPFIND, where the -b option didn't work.
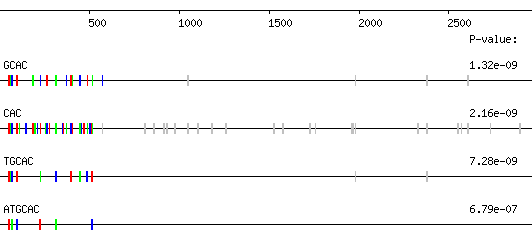
REPFIND produces a graphical display of the repeats that it finds, followed by a textual summary of each repeat cluster. An example output graphic, for the 3'UTR of the Xpat gene of the frog Xenopus laevis, is shown below. The bars show each position of the repeated word within the sequence; the colored bars indicate those repeats that form the strongest cluster. The different colors serve to distinguish repeats that are close together and have no meaning beyond that. On many computers you can save the image by right clicking on it and selecting the appropriate menu option. On other computers there should be other ways to do this.
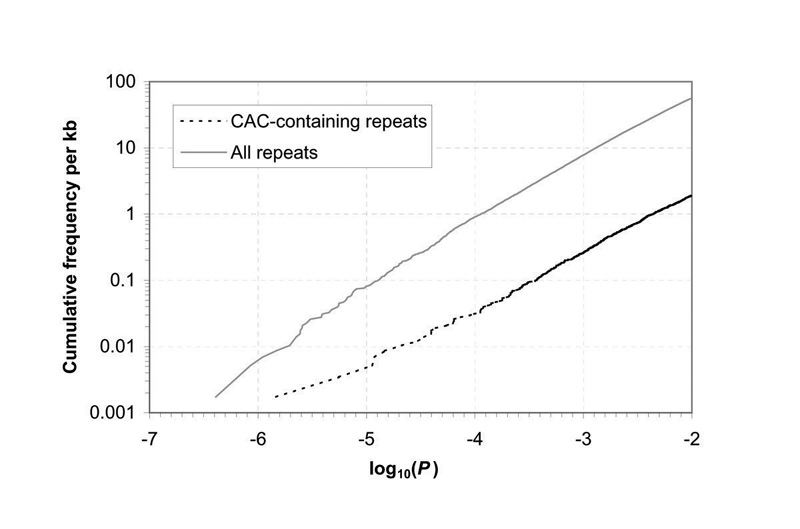
A P-value of 1e-05 means that such a concentration of that particular repeated word would be expected to occur by chance only one time in 10^5 clusters of the individual repeat being examined. Since REPFIND tests all possible words, we would like to know the frequency of any word achieving this P-value, that would be expected by chance in a random sequence. The following graph shows how often repeat clusters with particular P-values occur, per kb, in pseudorandom sequence. For example, a P-value of 1e-5 occurs on average once in 10,000 bp by chance.
This section describes some of the fields found on the REPFIND Input Form.
Digits, spaces, newlines, and fasta-style comments beginning with ">" will be ignored.
For example a GenBank accession number (e.g. X72340), an 'accession.version' number (e.g. X72340.1), or a GI number (e.g. 312302).
You may limit the search to a subsequence by entering its start and end coordinates. (The first nucleotide in the sequence has coordinate 1.) The default values of the From and To fields are the start and end of the sequence, respectively.
Only repeat clusters with P-values lower than this cutoff will be displayed.
Real nucleotide sequences often contain so-called low complexity sequence, meaning tracts of predominantly one nucleotide, dinucleotide tandem repeats, and the like. Since they probably do not correspond to the type of signal that you are looking for with REPFIND, they may be masked out with the program dust, which is widely used with other sequence analysis tools, such as BLAST, for similar reasons.
To calculate how unlikely a repeat is, REPFIND needs to know how abundant the nucleotides within the repeated word are. By default these abundances are obtained from the input sequence. However, they may also be obtained from databases of Xenopus, human, and S. cerevisiae 3' UTRs that we have compiled. By default, the abundances of dinucleotides are used, thus accounting for, e.g., reduced abundance of CpG relative to C and G. Alternatively, you may select the use of mononucleotides up to hexanucleotides, by selecting a Markov model of order zero to five. Since a 5th order Markov model requires frequencies of 4^6 = 4096 hexanucleotides, the dataset used for counting them should contain many more than this number of basepairs to get meaningful results.